Where the Deer and the Antelope Play
~ Laura Bashor Colorado State University
meta_data %>% filter(Host == 'Odocoileus virginianus') %>% group_by(Pango.lineage) %>% count() %>% rename(Count = n)
|
Count |
|
|
11 |
|
|
2 |
|
|
52 |
|
|
6 |
|
|
1 |
|
|
1 |
|
|
20 |
|
|
1 |
|
|
2 |
|
|
10 |
|
|
1 |
|
|
None |
1 |
df_deer <- meta_data %>% filter(Host == 'Odocoileus virginianus')
ggplot(df_deer) + geom_histogram(aes(x = Collection.date, fill = Pango.lineage)) + xlab('Collection Date') + ggtitle('Odocoileus virginianus 2020-09-28 to 2021-02-25')

temp <- df_deer %>% select(Location) %>% separate(Location, into = c('Region', 'Country', 'State'), sep = ' / ') %>% group_by(State) %>% count() %>% rename(Count = n)
|
State |
Count |
|
Iowa |
94 |
|
Ohio |
14 |

Mutations
mafft --retree 2 --maxiterate 1000 deer_seqs_filter.fasta > deer_aln.fasta
from Bio import SeqIO from Bio import SeqRecord from Bio import Seq with open('deer_aln_uc.fasta', 'w') as fd: for rec in SeqIO.parse('deer_aln.fasta', 'fasta'): seq = str(rec.seq).upper() new_seq = SeqRecord.SeqRecord(seq = Seq.Seq(seq), id = rec.id, description = rec.description) SeqIO.write(new_seq, fd, 'fasta')
python gisaid_mutations.py -i deer_aln_uc.fasta -o deer_mut.csv -g NC_045512.gb
gisaid_mutations.py compares aligned sequences against the the reference sequence. It uses the sequence NC_045512.2 GenBank annotation to identify genomic regions of interest.
The program identifies 537 nucleotide variations. Including 77 variations in the spike protein. You can see the genomic regions where the mutations were found in following table.
df_mut <- read.csv('output/deer_mut.csv') df_mut %>% group_by(product) %>% count() %>% rename(Count = n)
|
product |
Count |
|
36 |
|
|
2'-O-ribose
methyltransferase |
7 |
|
3'-to-5'
exonuclease |
35 |
|
3C-like
proteinase |
12 |
|
endoRNAse |
10 |
|
envelope
protein |
2 |
|
helicase |
26 |
|
leader
protein |
27 |
|
membrane
glycoprotein |
8 |
|
nsp10 |
6 |
|
nsp2 |
38 |
|
nsp3 |
62 |
|
nsp4 |
35 |
|
nsp6 |
16 |
|
nsp7 |
7 |
|
nsp8 |
10 |
|
nsp9 |
8 |
|
nucleocapsid
phosphoprotein |
33 |
|
ORF10
protein |
3 |
|
ORF3a
protein |
23 |
|
ORF7a
protein |
19 |
|
ORF7b |
1 |
|
ORF8
protein |
13 |
|
RNA-dependent
RNA polymerase |
23 |
|
surface
glycoprotein |
77 |
Here are the spike mutations.
df_mut %>% filter(product == 'surface glycoprotein') %>% select(reference.positions, ref_nucleotide, alt_nucleotide, codon, alt_codons, mutation)
|
reference.positions |
ref_nucleotide |
alt_nucleotide |
codon |
alt_codons |
mutation |
|
21575 |
C |
T |
CTT |
TTT |
L5F |
|
21597 |
C |
T |
TCT |
TTT |
S12F |
|
21614 |
C |
T |
CTT |
TTT |
L18F |
|
21618 |
C |
G |
ACA |
AGA |
T19R |
|
21622 |
C |
T |
ACC |
ACT |
T20T |
|
21627 |
C |
T |
ACT |
ATT |
T22I |
|
21648 |
C |
T |
ACT |
ATT |
T29I |
|
21658 |
C |
T |
TTC |
TTT |
F32F |
|
21705 |
T |
C |
TTA |
TCA |
L48S |
|
21707 |
C |
T |
CAT |
TAT |
H49Y |
|
21721 |
C |
T |
GAC |
GAT |
D53D |
|
21770 |
G |
C |
GTC |
CTC |
V70L |
|
21776 |
G |
A |
GGG |
AGG |
G72R |
|
21802 |
T |
C |
GAT |
GAC |
D80D |
|
21981 |
T |
- |
TTT |
T-T |
F140del |
|
21982 |
T |
- |
TTT |
TT- |
F140del |
|
21983 |
T |
- |
TTG |
-TG |
L141del |
|
21984 |
T |
- |
TTG |
T-G |
L141del |
|
21985 |
G |
- |
TTG |
TT- |
L141del |
|
21986 |
G |
- |
GGT |
-GT |
G142del |
|
21987 |
G |
- |
GGT |
G-T |
G142del |
|
21988 |
T |
- |
GGT |
GG- |
G142del |
|
21989 |
G |
- |
GTT |
-TT |
V143del |
|
21990 |
T |
- |
GTT |
G-T |
V143del |
|
21991 |
T |
- |
GTT |
GT- |
V143del |
|
21992 |
T |
- |
TAT |
-AT |
Y144del |
|
21993 |
A |
T |
TAT |
TTT |
Y144F |
|
22021 |
G |
T |
ATG |
ATT |
M153I |
|
22029 |
A |
- |
GAG |
G-G |
E156del |
|
22030 |
G |
- |
GAG |
GA- |
E156del |
|
22031 |
T |
- |
TTC |
-TC |
F157del |
|
22032 |
T |
- |
TTC |
T-C |
F157del |
|
22033 |
C |
- |
TTC |
TT- |
F157del |
|
22034 |
A |
- |
AGA |
-AG |
R158del |
|
22149 |
A |
- |
AAT |
A-T |
N196del |
|
22150 |
T |
- |
AAT |
AA- |
N196del |
|
22162 |
T |
C |
TAT |
TAC |
Y200Y |
|
22295 |
C |
T |
CAT |
TAT |
|
|
22350 |
C |
T |
GCA |
GTA |
A263V |
|
22432 |
C |
T |
GAC |
GAT |
D290D |
|
22450 |
C |
T |
CTC |
CTT |
L296L |
|
22498 |
C |
T |
ATC |
ATT |
I312I |
|
22627 |
G |
A |
AGG |
AGA |
R355R |
|
22669 |
T |
C |
TAT |
TAC |
Y369Y |
|
22687 |
C |
T |
TCC |
TCT |
S375S |
|
22750 |
T |
C |
TAT |
TAC |
Y396Y |
|
22798 |
A |
G |
CCA |
CCG |
P412P |
|
22995 |
C |
A |
ACA |
AAA |
|
|
23014 |
A |
C |
GAA |
GAC |
|
|
23066 |
G |
T |
GGT |
TGT |
G502C |
|
23266 |
C |
T |
GAC |
GAT |
D568D |
|
23277 |
C |
T |
ACT |
ATT |
T572I |
|
23282 |
G |
T |
GAT |
TAT |
D574Y |
|
23284 |
T |
C |
GAT |
GAC |
D574D |
|
23401 |
G |
T |
CAG |
CAT |
Q613H |
|
23403 |
A |
G |
GAT |
GGT |
|
|
23604 |
C |
G |
CCT |
CGT |
|
|
23664 |
C |
T |
GCA |
GTA |
|
|
23740 |
T |
C |
ATT |
ATC |
I726I |
|
24055 |
T |
C |
GCT |
GCC |
A831A |
|
24206 |
A |
T |
ATC |
TTC |
I882F |
|
24232 |
A |
G |
GCA |
GCG |
A890A |
|
24320 |
C |
A |
CAA |
AAA |
Q920K |
|
24337 |
C |
T |
AAC |
AAT |
N925N |
|
24410 |
G |
A |
GAT |
AAT |
|
|
24418 |
C |
A |
GTC |
GTA |
V952V |
|
24542 |
G |
T |
GAT |
TAT |
D994Y |
|
24544 |
T |
C |
GAT |
GAC |
D994D |
|
24781 |
G |
A |
AAG |
AAA |
K1073K |
|
24900 |
A |
- |
CAA |
C-A |
Q1113del |
|
24997 |
A |
G |
TTA |
TTG |
L1145L |
|
24998 |
G |
T |
GAC |
TAC |
D1146Y |
|
25018 |
A |
G |
TTA |
TTG |
L1152L |
|
25169 |
C |
T |
CTT |
TTT |
|
|
25244 |
G |
T |
GTA |
TTA |
|
|
25292 |
C |
A |
CTC |
ATC |
L1244I |
|
25350 |
C |
A |
CCA |
CAA |
P1263Q |
Mutual Information
python gisaid_MI.py -i deer_aln_uc.fasta -o deer_mi.csv
|
Position_1 |
Position_2 |
MI |
|
10319 |
25907 |
0.941045455 |
|
25907 |
28472 |
0.883140787 |
|
25907 |
27964 |
0.844033431 |
|
27964 |
28472 |
0.816303445 |
|
10319 |
28472 |
0.816298105 |
|
10319 |
27964 |
0.768796929 |
|
204 |
24997 |
0.764232905 |
|
12525 |
13665 |
0.74484856 |
|
11919 |
12737 |
0.729264896 |
|
13665 |
28893 |
0.728748237 |
|
12525 |
28893 |
0.720429105 |
|
11195 |
11919 |
0.714923206 |
|
19180 |
21776 |
0.711697689 |
|
12737 |
12792 |
0.711413545 |
|
11195 |
12792 |
0.704167711 |
|
18424 |
25907 |
0.701801109 |
|
10319 |
18424 |
0.701731398 |
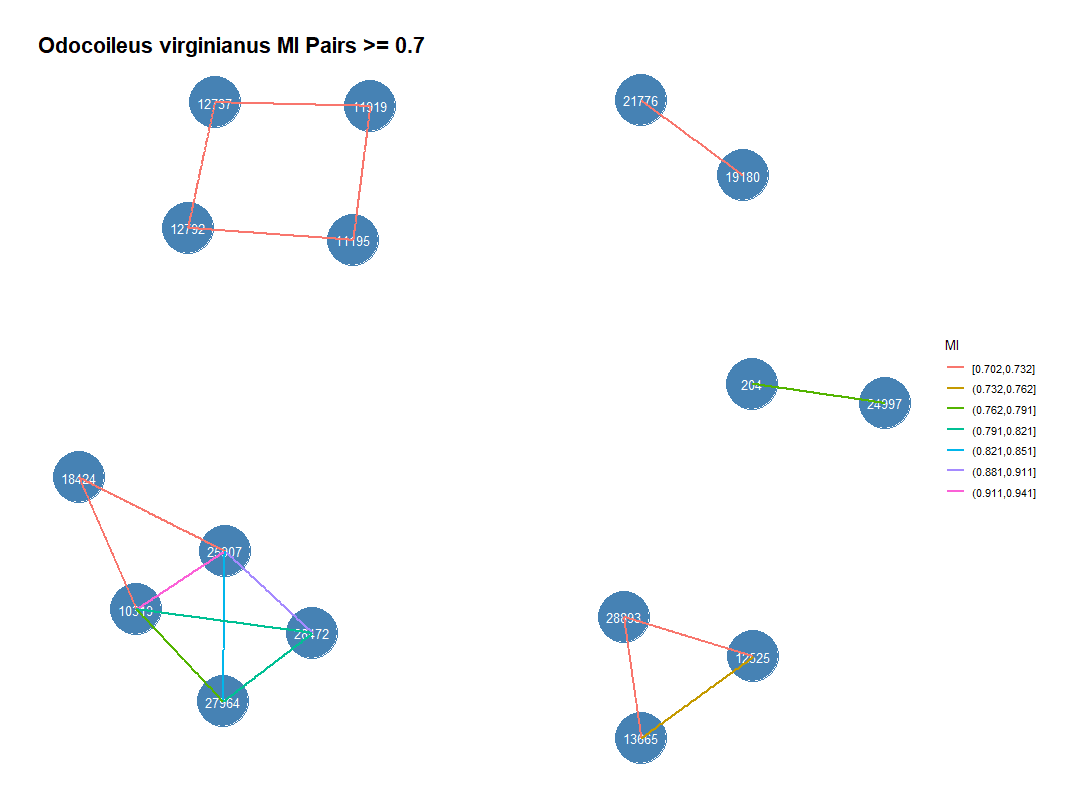
python gisaid_MI_features.py -i deer_mi.csv -o deer_mi_features.csv -m deer_mut.csv -c 0.7
|
ref.positions |
ref_nucleotide |
alt_nucleotide |
codon |
alt_codons |
mutation |
product |
|
10319 |
C |
T |
CTT |
TTT |
L89F |
3C-like
proteinase |
|
25907 |
G |
T |
GGT |
GTT |
G172V |
ORF3a
protein |
|
28472 |
C |
T |
CCT |
TCT |
P67S |
nucleocapsid
phosphoprotein |
|
27964 |
C |
T |
TCA |
TTA |
S24L |
ORF8
protein |
|
204 |
G |
T |
||||
|
24997 |
A |
G |
TTA |
TTG |
L1145L |
surface
glycoprotein |
|
12525 |
C |
T |
ACA |
ATA |
T145I |
nsp8 |
|
13665 |
C |
T |
CAC |
CAT |
H66H |
RNA-dependent
RNA polymerase |
|
11919 |
C |
T |
TCT |
TTT |
S26F |
nsp7 |
|
12737 |
A |
G |
ACT |
GCT |
T18A |
nsp9 |
|
28893 |
C |
T |
CCT |
CTT |
P207L |
nucleocapsid
phosphoprotein |
|
11195 |
C |
T |
CTT |
TTT |
L75F |
nsp6 |
|
19180 |
G |
T |
GTA |
TTA |
V381L |
3'-to-5'
exonuclease |
|
21776 |
G |
A |
GGG |
AGG |
G72R |
surface
glycoprotein |
|
12792 |
A |
T |
AAG |
ATG |
K36M |
nsp9 |
|
18424 |
A |
G |
AAT |
GAT |
N129D |
3'-to-5'
exonuclease |


Comments
Post a Comment